Description
Predicting protein functional association in microbial and viral genomes by analysis of conservation of genomic context.
Functional associations of proteins can be predicted by conservation of the genomic neighbourhood surrounding the gene encoding the protein of interest. Our tool FlaGs clusters neighbourhood-encoded proteins into homologous groups and outputs the identity of the groups, a graphical visualization of the gene neighbourhood and its conservation, and optionally, a phylogenetic tree annotated with flanking gene conservation.
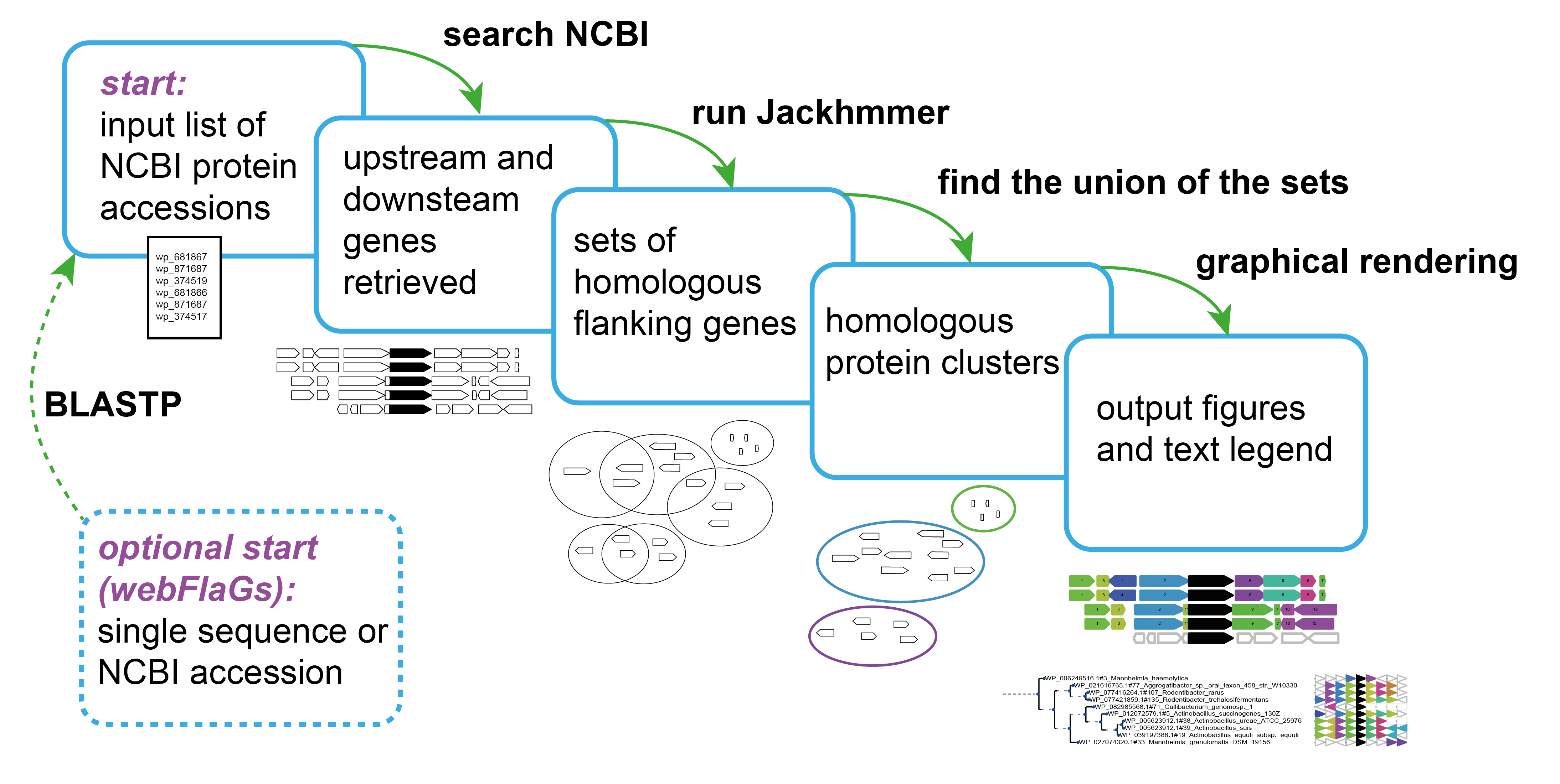
Source code and documentation are available at the GitHub Page.
Output examples


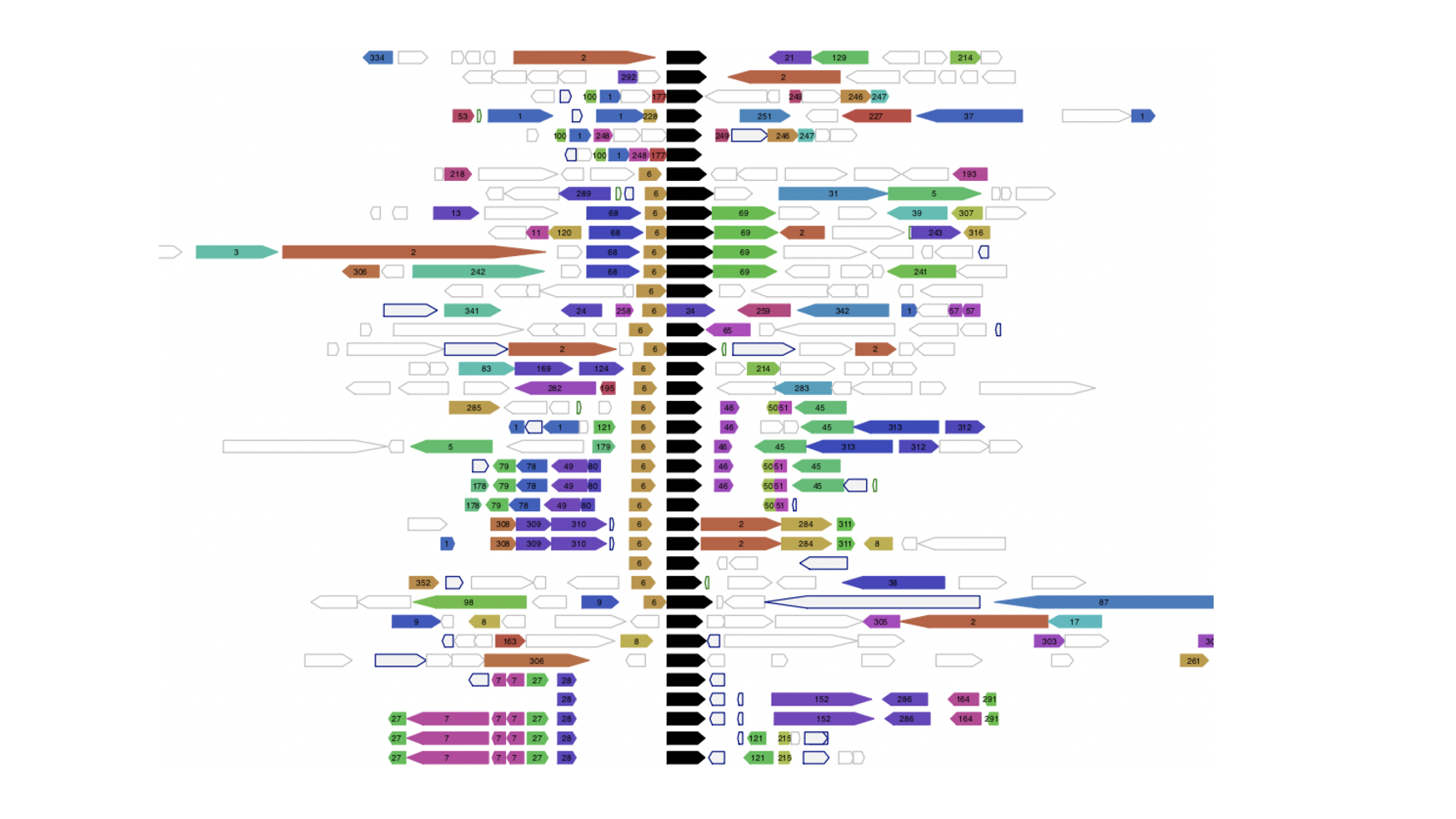
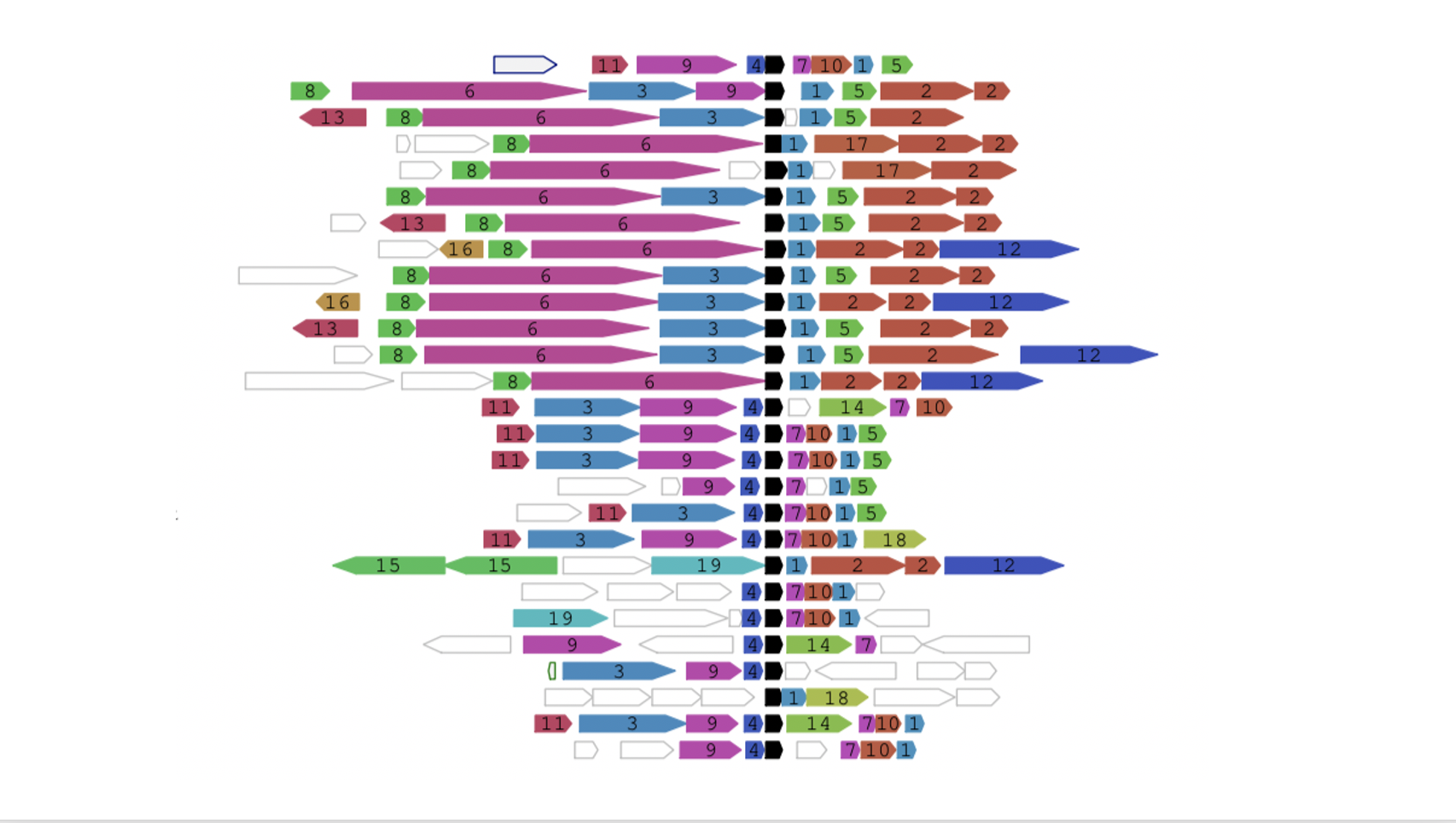
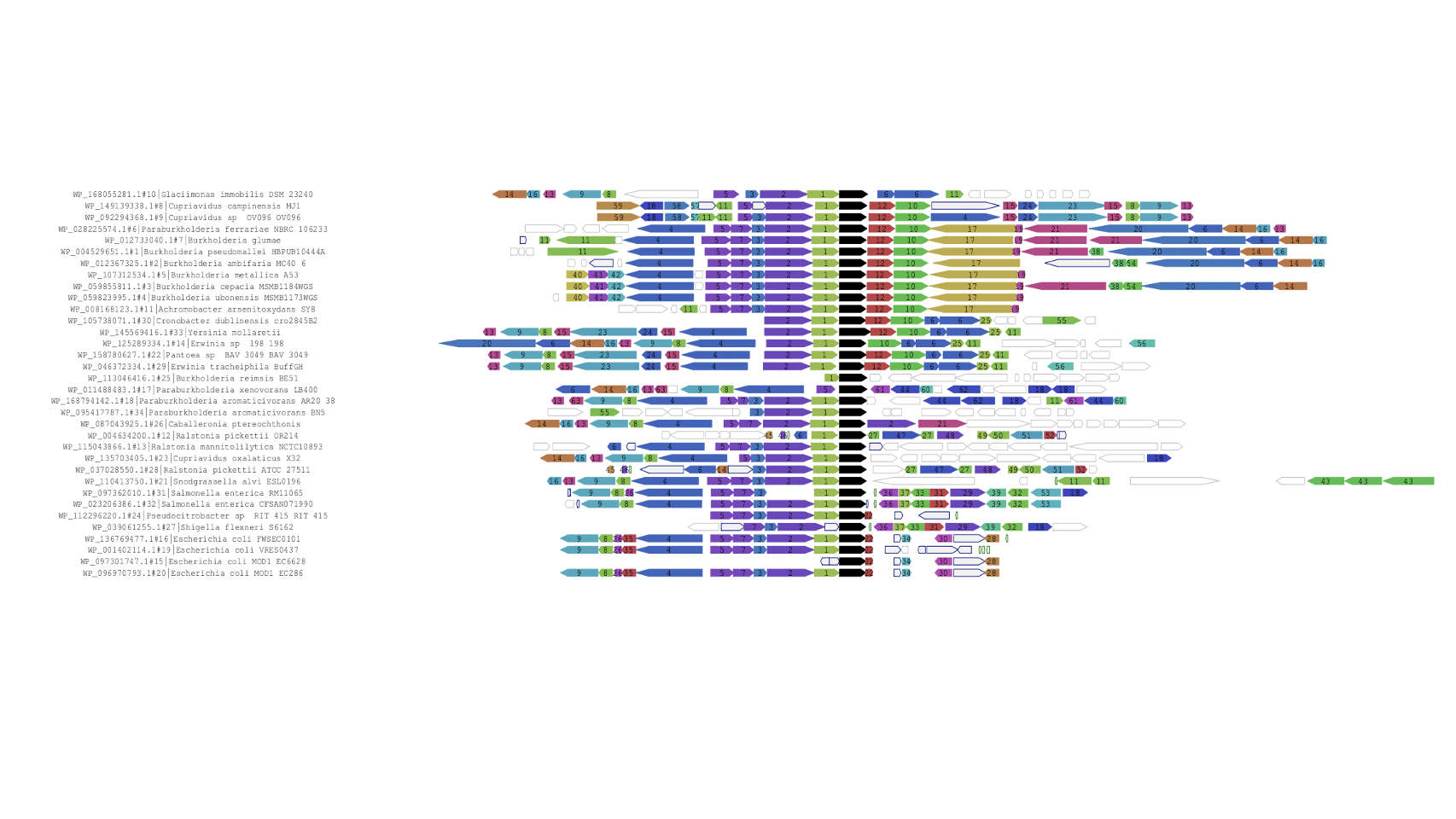
Reference
If you find webFlaGs useful, please cite:
Chayan Kumar Saha, Rodrigo Sanches Pires, Harald Brolin, Maxence Delannoy,
Gemma Catherine Atkinson, FlaGs and webFlaGs: discovering novel
biology through the analysis of gene neighbourhood conservation,
Bioinformatics, Volume 37, Issue 9, 1 May 2021, Pages 1312–1314;
doi:10.1093/bioinformatics/btaa788
Authors & Contact
FlaGs was first made by Chayan Kumar Saha and Gemma C. Atkinson. It is now
developed by the Atkinson FlaGs team: Jose Nakamoto, Artyom Egorov and Veda Bojar at the
Department of Experimental Medical Science, Lund University, Sweden.
We are open for suggestions of how we can extend and improve webFlaGs functionality. Please don't hesitate to share your ideas or feature requests.
Please contact us by e-mail or use GitHub Issues to report any technical problems related to FlaGs.
Submission form
You can try an example with a set of proteins.
For creating an input file for FlaGs from the results of an online BlastP or PSI-Blast search at the NCBI, you can use this guideline.