Description
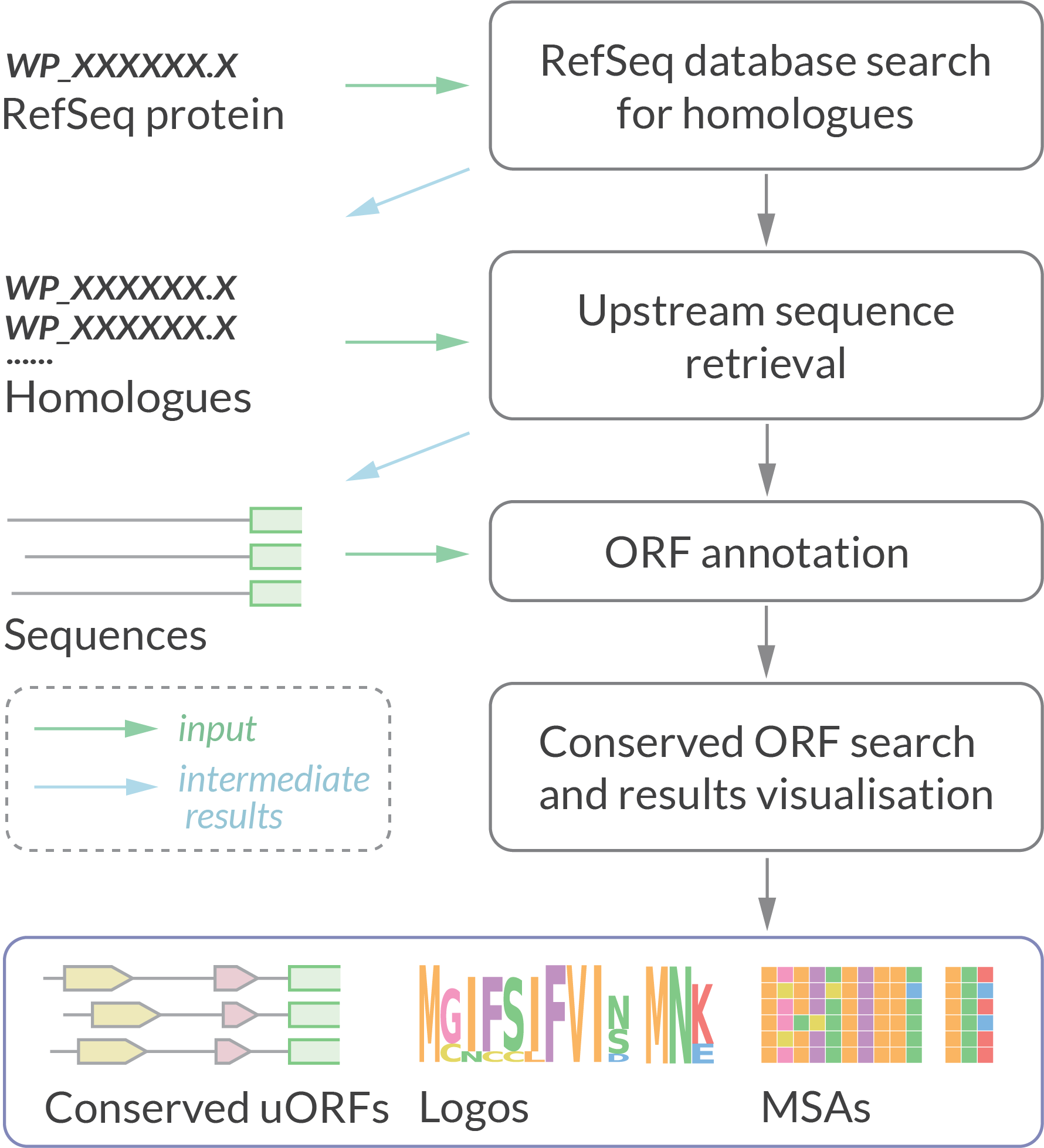
uORF4u is a bioinformatics tool for conserved uORF annotation in 5′ upstream sequences of a user-defined protein of interest or a set of protein homologues. It can also be used to find small ORFs within a set of nucleotide sequences.
The output includes publication-quality figures with multiple sequence alignments, sequence logos and locus annotation of the predicted uORFs in graphical vector format.
Source code available at the GitHub Page.
We also recommend visiting the tool's detailed documentation website that provides an example-driven guide and documentation to the command-line version and python API.
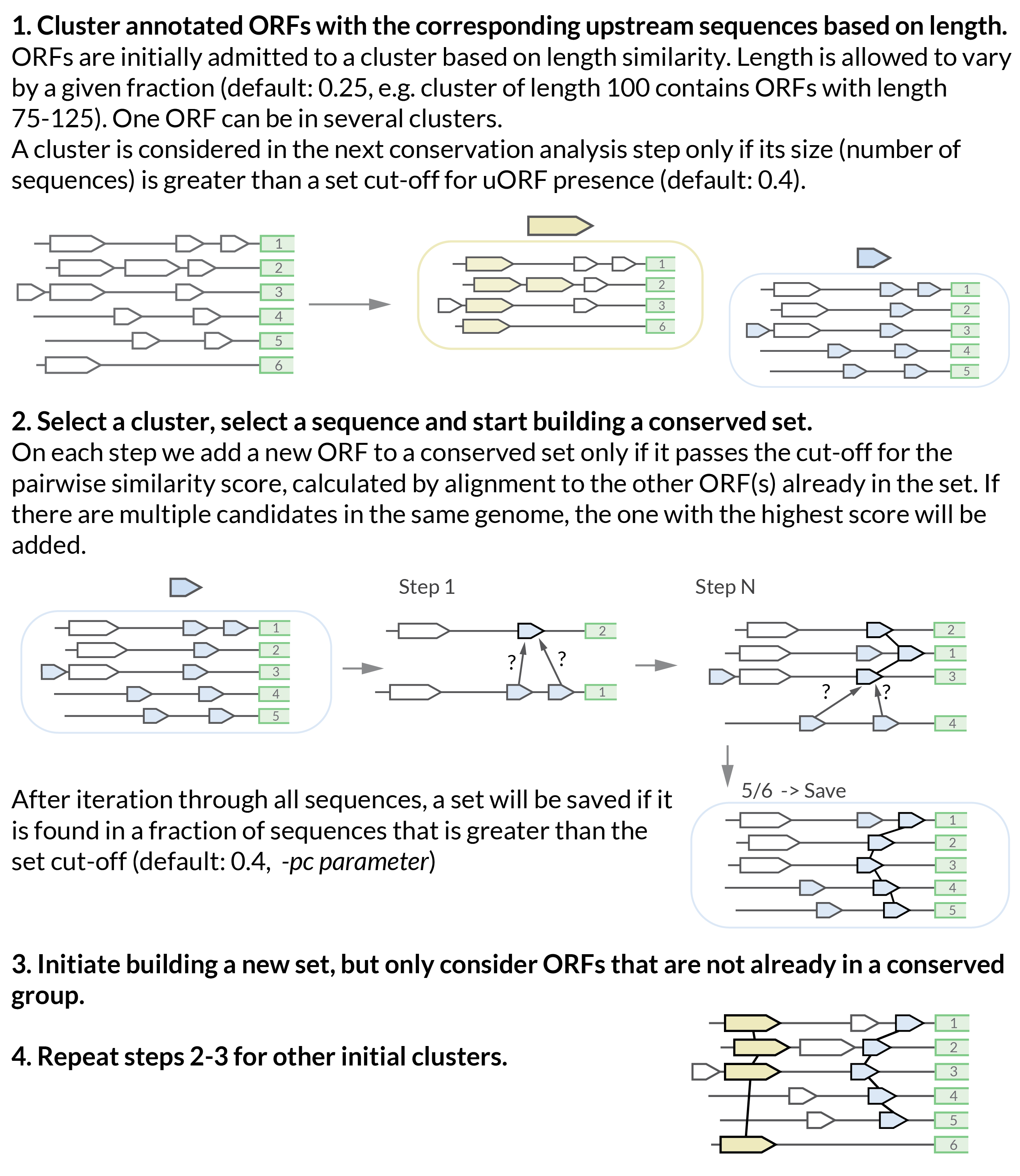
How does the conservation analysis algorithm work?
Reference
If you find uorf4u useful, please, cite:
Artyom A. Egorov, Gemma C. Atkinson, uORF4u: a tool for annotation
of conserved upstream open reading frames
Bioinformatics, Volume 39, Issue 5, May 2023, btad323; doi:
10.1093/bioinformatics/btad323
Authors & Contact
uORF4u is developed by Artyom Egorov at the Atkinson Lab, Department of
Experimental Medical Science, Lund University, Sweden.
We are open for suggestions of how we can extend and improve uorf4u functionality. Please
don't hesitate to share your ideas or
feature requests.
Please contact us by e-mail or use GitHub Issues to report any technical problems related to uORF4u. You can also use Discussions section for sharing your ideas or feature requests!
Submission form
You can try an example with list of 6 ErmC homologues.